Level and plasticity divergence during nitrogen starvation
source("functions_for_figure_scripts.R")
load("data_files/FinalDataframe3Disp.RData")
load("data_files/Cleaned_Counts.RData")
load("data_files/Cleaned_Counts_Allele.RData")
# change these variables for different environment-specific scripts:
experiment_name <- "LowN"
clusterdf_name <- "LowN_2"
expdf <- filter(finaldf, experiment == experiment_name)
folder_name <- "LowNitrogen"Summary of number of genes per divergence category:
# summary of groups in this experiment
table(expdf$group) |> sort(decreasing = TRUE)##
## cons1 levupcer1 cons0 cons2
## 1082 585 455 441
## dyn01 dyn10 levuppar1 levdynupcer10
## 363 252 190 124
## levuppar0 levuppar2 levdynuppar01 levupcer0
## 110 105 96 94
## dyn20 levdynupcer01 dyn02 dyn21
## 88 79 62 50
## levupcer2 levdynuppar02 levdynuppar10 levdynupcer20
## 45 39 32 30
## dyn12 levdynupcer21 levdynuppar12 levdynuppar20
## 29 18 13 11
## levdynupcer12 levdynuppar21 levdynupcer02
## 10 7 2sum(table(expdf$group))## [1] 4412length(unique(expdf$group))## [1] 27code_order <- names(sort(table(expdf$group), decreasing = TRUE))Fisher exact test for group enrichment
# fisher exact test for un-enrichment of plasticity divergence
# among level divergent set
plotdf <- expdf |> filter(level != "low_expr")
n_leveldiv <- sum(plotdf$level == "diverged")
n_levelcons <- sum(plotdf$level == "conserved")
n_dyndiv <- sum(plotdf$plasticity == "diverged")
n_dyncons <- sum(plotdf$plasticity == "conserved")
n_dynandlevdiv <- sum(plotdf$plasticity == "diverged" &
plotdf$level == "diverged")
fisher.test(matrix(c(n_dynandlevdiv, n_dyndiv - n_dynandlevdiv,
n_leveldiv - n_dynandlevdiv,
n_dyncons - (n_leveldiv - n_dynandlevdiv)),
nrow = 2))##
## Fisher's Exact Test for Count Data
##
## data: matrix(c(n_dynandlevdiv, n_dyndiv - n_dynandlevdiv, n_leveldiv - n_dynandlevdiv, n_dyncons - (n_leveldiv - n_dynandlevdiv)), nrow = 2)
## p-value = 0.5364
## alternative hypothesis: true odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.833891 1.097546
## sample estimates:
## odds ratio
## 0.9569531# barplot of all divergence groups
ggplot(expdf, aes(x = group)) +
geom_bar(aes(fill = group4)) +
scale_x_discrete(breaks = code_order, limits = code_order) +
scale_fill_discrete(type = colordf[colordf$scheme == "group4",]$type,
labels = colordf[colordf$scheme == "group4",]$labels,
limits = colordf[colordf$scheme == "group4",]$limits) +
theme_classic() +
theme(axis.text.x = element_text(angle = 90),
legend.position = "none") +
geom_text(stat='count', aes(label=after_stat(count)), vjust=-1) +
ylim(c(0, 2750))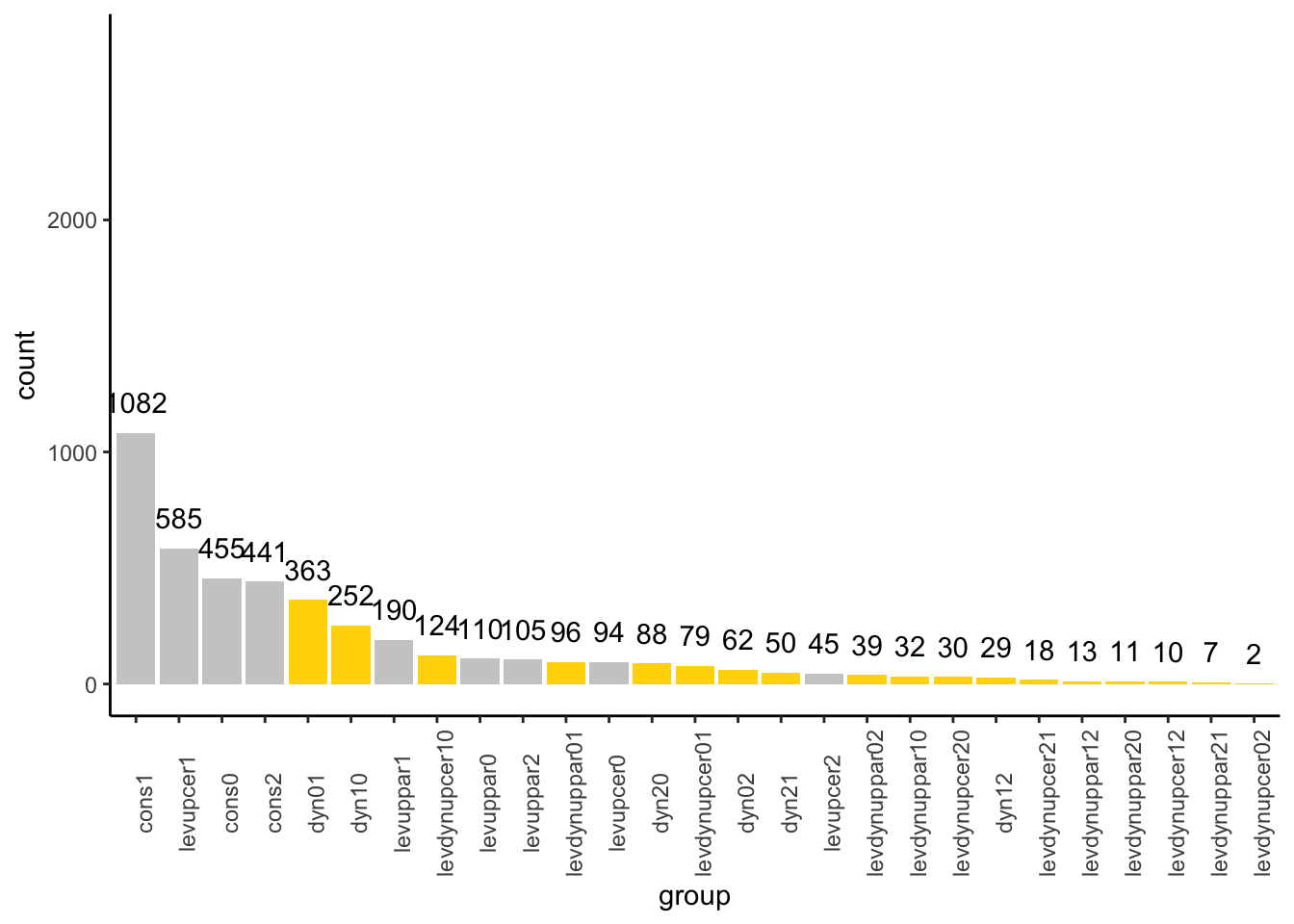
Gene count tables
# Biased genes, can't determine level divergence:
expdf |> filter(level == "biased") |> nrow()## [1] 83# Lowly expressed genes in this environment:
setdiff(unique(finaldf$gene_name), expdf$gene_name) |> length()## [1] 451# Scer gene counts:
expdf |> select(cer) |> table()## cer
## 0 1 2
## 1300 2317 795expdf |> select(par) |> table()## par
## 0 1 2
## 1196 2470 746# Conserved genes:
expdf |> filter(plasticity != "diverged" &
level != "diverged") |>
select(cer, par) |> table()## par
## cer 0 1 2
## 0 455 0 0
## 1 0 1082 0
## 2 0 0 441# Diverged level:
expdf |> filter(plasticity != "diverged" &
level == "diverged") |>
mutate(direction = if_else(effect_size_species > 0,
true = "up in Scer",
false = "up in Spar")) |>
select(cer, par, direction) |> table()## , , direction = up in Scer
##
## par
## cer 0 1 2
## 0 94 0 0
## 1 0 585 0
## 2 0 0 45
##
## , , direction = up in Spar
##
## par
## cer 0 1 2
## 0 110 0 0
## 1 0 190 0
## 2 0 0 105# Diverged plasticity:
expdf |> filter(plasticity == "diverged" &
level != "diverged") |>
mutate(isZero = if_else(cer == 0 | par == 0,
true = if_else(cer == 0,
true = "Scer0",
false = "Spar0"),
false = "neither0")) |>
select(cer, par, isZero) |> table()## , , isZero = neither0
##
## par
## cer 0 1 2
## 0 0 0 0
## 1 0 0 29
## 2 0 50 0
##
## , , isZero = Scer0
##
## par
## cer 0 1 2
## 0 0 363 62
## 1 0 0 0
## 2 0 0 0
##
## , , isZero = Spar0
##
## par
## cer 0 1 2
## 0 0 0 0
## 1 252 0 0
## 2 88 0 0# Diverged level and plasticity:
expdf |> filter(plasticity == "diverged" &
level == "diverged") |>
mutate(direction = if_else(effect_size_species > 0,
true = "up in Scer",
false = "up in Spar")) |>
select(cer, par, direction) |> table()## , , direction = up in Scer
##
## par
## cer 0 1 2
## 0 0 79 2
## 1 124 0 10
## 2 30 18 0
##
## , , direction = up in Spar
##
## par
## cer 0 1 2
## 0 0 96 39
## 1 32 0 13
## 2 11 7 0Average expression of genes in each divergence category
Genes with conserved level and plasticity
# Conserved genes:
ylims <- c(3, 9)
cluster_names <- expdf |> filter(plasticity != "diverged" &
level != "diverged") |>
select(cer, par) |> pull() |> unique()
plotlist <- vector(mode = "list", length = length(cluster_names))
names(plotlist) <- cluster_names
for (clust in cluster_names) {
gene_idxs <- expdf |> filter(plasticity != "diverged" &
level != "diverged" &
cer == clust &
par == clust) |>
select(gene_name) |> pull()
plotlist[[as.character(clust)]] <- annotate_figure(plotGenes(.gene_idxs = gene_idxs, .quartet = FALSE,
.experiment_name = experiment_name,
.plotlims = ylims) +
theme(plot.margin = unit(c(0,0.25,0,0), "cm")),
top = paste0(length(gene_idxs), " genes"))
}## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`### Main figure: example gene group
pdf(file = paste0("paper_figures/",
folder_name, "/conserved.pdf"), width = 2, height = 2)
plotlist$`1`
dev.off()## quartz_off_screen
## 2# ### Supplement: top 3 gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/conserved_top3.pdf"), width = 2, height = 5)
# ggarrange(plotlist = plotlist[1:3], nrow = 3, ncol = 1)
# dev.off()
# ### Supplement: all gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/conserved_all.pdf"), width = 2, height = (5/3)*length(plotlist))
# ggarrange(plotlist = plotlist, nrow = length(plotlist), ncol = 1)
# dev.off()Genes with conserved plasticity and diverged level
# Diverged level:
griddf <- expdf |> filter(plasticity != "diverged" &
level == "diverged") |>
mutate(lfc_sign = sign(effect_size_species)) |>
group_by(cer, par, lfc_sign) |> summarise(n = n()) |>
arrange(desc(n))## `summarise()` has grouped output by 'cer', 'par'. You
## can override using the `.groups` argument.plotlist <- vector(mode = "list", length = 0)
for (i in c(1:nrow(griddf))) {
clust <- griddf$cer[i]
lfcsign <- griddf$lfc_sign[i]
gene_idxs <- expdf |> filter(plasticity != "diverged" &
level == "diverged" &
cer == clust &
par == clust &
sign(effect_size_species) == lfcsign) |>
select(gene_name) |> pull()
plotlist[[paste0(clust, lfcsign)]] <- annotate_figure(plotGenes(.gene_idxs = gene_idxs, .quartet = FALSE,
.experiment_name = experiment_name,
.plotlims = ylims) +
theme(plot.margin = unit(c(0,0.25,0,0), "cm")),
top = paste0(length(gene_idxs), " genes"))
}## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`## Warning: Removed 1 row containing missing values or values
## outside the scale range (`geom_line()`).## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`### Main figure: example gene group
pdf(file = paste0("paper_figures/",
folder_name, "/level.pdf"), width = 2, height = 2)
plotlist$`11`
dev.off()## quartz_off_screen
## 2# ### Supplement: top 3 gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/level_top3.pdf"), width = 2, height = 5)
# ggarrange(plotlist = plotlist[1:3], nrow = 3, ncol = 1)
# dev.off()
# ### Supplement: all gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/level_all.pdf"), width = 2, height = (5/3)*length(plotlist))
# ggarrange(plotlist = plotlist, nrow = length(plotlist), ncol = 1)
# dev.off()Genes with diverged plasticity and conserved level
griddf <- expdf |> filter(plasticity == "diverged" &
level != "diverged") |>
group_by(cer, par) |>
summarise(n = n()) |>
arrange(desc(n))## `summarise()` has grouped output by 'cer'. You can
## override using the `.groups` argument.plotlist <- vector(mode = "list", length = 0)
for (i in c(1:nrow(griddf))) {
clust_cer <- griddf$cer[i]
clust_par <- griddf$par[i]
gene_idxs <- expdf |> filter(plasticity == "diverged" &
level != "diverged" &
cer == clust_cer &
par == clust_par) |>
select(gene_name) |> pull()
plotlist[[paste0(clust_cer, clust_par)]] <- annotate_figure(plotGenes(.gene_idxs = gene_idxs, .quartet = FALSE,
.experiment_name = experiment_name,
.plotlims = ylims) +
theme(plot.margin = unit(c(0,0.25,0,0), "cm")),
top = paste0(length(gene_idxs), " genes"))
}## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`### Main figure: example gene group
pdf(file = paste0("paper_figures/",
folder_name, "/plasticity.pdf"), width = 2, height = 2)
plotlist$`21`
dev.off()## quartz_off_screen
## 2# ### Supplement: top 3 gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/plasticity_top3.pdf"), width = 2, height = 5)
# ggarrange(plotlist = plotlist[1:3], nrow = 3, ncol = 1)
# dev.off()
# ### Supplement: all gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/plasticity_all.pdf"), width = 2, height = (5/3)*length(plotlist))
# ggarrange(plotlist = plotlist, nrow = length(plotlist), ncol = 1)
# dev.off()Genes with diverged plasticity and diverged level
# Diverged level and plasticity:
griddf <- expdf |> filter(plasticity == "diverged" &
level == "diverged") |>
mutate(lfc_sign = sign(effect_size_species)) |>
group_by(cer, par, lfc_sign) |>
summarise(n = n()) |>
arrange(desc(n))## `summarise()` has grouped output by 'cer', 'par'. You
## can override using the `.groups` argument.plotlist <- vector(mode = "list", length = 0)
for (i in c(1:nrow(griddf))) {
clust_cer <- griddf$cer[i]
clust_par <- griddf$par[i]
lfcsign <- griddf$lfc_sign[i]
gene_idxs <- expdf |> filter(plasticity == "diverged" &
level == "diverged" &
cer == clust_cer &
par == clust_par &
sign(effect_size_species) == lfcsign) |>
select(gene_name) |> pull()
plotlist[[paste0(clust_cer, clust_par, lfcsign)]] <- annotate_figure(plotGenes(.gene_idxs = gene_idxs, .quartet = FALSE,
.experiment_name = experiment_name,
.plotlims = ylims) +
theme(plot.margin = unit(c(0,0.25,0,0), "cm")),
top = paste0(length(gene_idxs), " genes"))
}## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## `summarise()` has grouped output by 'group_id',
## 'gene_name', 'experiment'. You can override using the
## `.groups` argument.
## `summarise()` has grouped output by 'time_point_num',
## 'experiment'. You can override using the `.groups`
## argument.
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`
## Adding missing grouping variables: `time_point_num`,
## `experiment`### Main figure: example gene group
pdf(file = paste0("paper_figures/",
folder_name, "/level_and_plasticity.pdf"), width = 2, height = 2)
plotlist$`01-1`
dev.off()## quartz_off_screen
## 2# ### Supplement: top 3 gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/level_and_plasticity_top3.pdf"), width = 2, height = 5)
# ggarrange(plotlist = plotlist[1:3], nrow = 3, ncol = 1)
# dev.off()
# ### Supplement: all gene groups
# pdf(file = paste0("paper_figures/Supplement/",
# folder_name, "/level_and_plasticity_all.pdf"), width = 2, height = (5/3)*length(plotlist))
# ggarrange(plotlist = plotlist, nrow = length(plotlist), ncol = 1)
# dev.off()Proportional area plot of gene groups
### Proportional area plot for Main figure
prop_table <- expdf |> mutate(plasticity_diverged = plasticity == "diverged",
level_diverged = level == "diverged") |>
select(plasticity_diverged, level_diverged) |> table()
prop_table## level_diverged
## plasticity_diverged FALSE TRUE
## FALSE 1978 1129
## TRUE 844 461sum(prop_table)## [1] 4412prop_table/sum(prop_table)## level_diverged
## plasticity_diverged FALSE TRUE
## FALSE 0.4483228 0.2558930
## TRUE 0.1912965 0.1044878pdf(file = paste0("paper_figures/",
folder_name, "/prop_area.pdf"), width = 4, height = 4)
plotPropAreaSingle(.counts = c(prop_table[1, 2], prop_table[1, 1],
prop_table[2, 1], prop_table[2, 2]),
.colors = colordf[colordf$scheme == "group4",]$type[c(3, 1, 2, 4)],
.patterns = colordf[colordf$scheme == "group4",]$pattern[c(3, 1, 2, 4)],
.text_size = 10, .text_bounds = 20)
dev.off()## quartz_off_screen
## 2Choosing example level-diverging and plasticity-diverging gene for workflow figure
### Choosing example genes for LFC calculation figure panel
if (experiment_name == "HAP4") {
# choosing one level-diverging and one plasticity-diverging gene
# level
expdf |> filter(level == "diverged" & plasticity == "conserved") |> select(gene_name) |>
pull() |> sample(1)
# we chose: YBR067C
pdf("paper_figures/ExperimentOverview/level_expr.pdf",
width = 2, height = 2)
p <- plotGenes("YBR067C", .experiment_name = experiment_name,
.normalization = "log2")
print(annotate_figure(p, top = "YBR067C"))
dev.off()
# plasticity
expdf |> filter(level == "conserved" & plasticity == "diverged") |> select(gene_name) |>
pull() |> sample(1) # YNL312W is absolutely wild, nearly mirror image
# We chose: YJR001W
pdf("paper_figures/ExperimentOverview/plasticity_expr.pdf",
width = 2, height = 2)
p <- plotGenes("YJR001W", .experiment_name = experiment_name, .normalization = "log2")
print(annotate_figure(p, top = "YJR001W"))
dev.off()
}Boxplots of level divergence in each plasticity category
Do diverging-plasticity genes have stronger log2 fold change than conserved plasticity genes? We’ve looked for group enrichment with the Fisher’s Exact test, but perhaps we missed a more subtle relationship between the magnitude of log2 fold change and plasticity divergence.
p_thresh <- 0.05
plotdf <- expdf |>
filter(padj_species < p_thresh) |> # only significant level divergers
mutate(lfc_direction = sign(effect_size_species)) |>
arrange(plasticity, lfc_direction, cer, par)
cluster_levels <- unique(paste0(plotdf$cer, plotdf$par, plotdf$lfc_direction))
plotdf$cluster_group <- paste0(plotdf$cer, plotdf$par, plotdf$lfc_direction) |>
factor(levels = cluster_levels)
# t-tests of upScer and upSpar log2 fold changes
# upScer divergent vs conserved plasticity genes
cons_fold_changes <- plotdf |> filter(plasticity == "conserved" &
lfc_direction == 1) |>
select(effect_size_species) |> pull()
div_fold_changes <- plotdf |> filter(plasticity == "diverged" &
lfc_direction == 1) |>
select(effect_size_species) |> pull()
p_upcer <- t.test(x = cons_fold_changes, y = div_fold_changes)$p.value
# upSpar divergent vs conserved plasticity genes
cons_fold_changes <- plotdf |> filter(plasticity == "conserved" &
lfc_direction == -1) |>
select(effect_size_species) |> pull()
div_fold_changes <- plotdf |> filter(plasticity == "diverged" &
lfc_direction == -1) |>
select(effect_size_species) |> pull()
p_uppar <- t.test(x = cons_fold_changes, y = div_fold_changes)$p.value
# scatter plot of each cluster combination to screen
# for clusters with more level divergence than others
# only sig level-diverging genes
p <- ggplot(plotdf, aes(x = cluster_group,
y = effect_size_species)) +
geom_boxplot(aes(color = interaction(lfc_direction, plasticity))) +
theme_classic() +
ylab("log2 fold change") +
xlab("plasticity cluster") +
scale_x_discrete(limits = cluster_levels, drop = FALSE) +
geom_hline(yintercept = 0) +
ylim(c(-11, 11)) +
geom_text(x = 10, y = 10, label = paste0("p = ", round(p_upcer, digits = 2))) +
geom_text(x = 5, y = -10, label = paste0("p = ", round(p_uppar, digits = 2))) +
theme(legend.title = element_blank())
pdf(paste0("paper_figures/Supplement/supp_levdyn_boxplots",
experiment_name, ".pdf"),
width = 7, height = 4)
p
dev.off()## quartz_off_screen
## 2